Stichprobebasierte Quantendiagonalisierung vomme Chemie-Hamiltonian
Nutzungsschätzung: unter einer Minute auf emm Heron r2 Prozessor (HINWEIS: Des isch bloß a Schätzung. Ihre Laufzeit ka variiere.)
Hintergrund
In dem Tutorial zeige mir, wie verrauschte Quantenstichproben nachbearbeitet werde, um a Approximation vom Grundzustand vom Stickstoffmolekül bei Gleichgewichtsbindungslänge z'finde. Dabei verwende mir den stichprobebasierte Quantendiagonalisierungsalgorithmus (SQD) für Stichproben aus emm 59-Qubit-Quantenschaltkreis (52 System-Qubits + 7 Ancilla-Qubits). A Qiskit-basierte Implementierung isch verfügbar in denne SQD Qiskit Addons, weitere Details findet Se in der entsprechende Dokumentation mit emm einfache Beispiel zum Einstieg.
SQD isch a Technik zum Finde von Eigenwert und Eigenvektore von Quantenoperatore, wie dem Hamiltonian vomme Quantensystem, unter Verwendung von Quanten- und verteiltem klassischem Computing mitanand. Des verteilte klassische Computing wird verwendet, um Stichproben von emm Quantenprozessor z'verarbeite und an Ziel-Hamiltonian in an durch sie aufgespannte Unterraum z'projiziere und z'diagonalisiere. Des ermöglicht SQD, robust gegenüber durch Quantenrauschen verfälschte Stichproben z'sei und mit große Hamiltonians umz'geh, wie Chemie-Hamiltonians mit Millione von Wechselwirkungstermen, die jenseits der Reichweite exakter Diagonalisierungsmethode liege. An SQD-basierte Workflow hat folgende Schritt:
- Wähle Se an Schaltkreis-Ansatz und wende Se ihn auf emm Quantencomputer auf an Referenzzustand an (in dem Fall den Hartree-Fock-Zustand).
- Sampele Se Bitstrings aus dem resultierende Quantenzustand.
- Führe Se des selbstkonsistente Konfigurationswiederherstellungsverfahre auf denne Bitstrings aus, um die Approximation vom Grundzustand z'erhalte.
SQD funktioniert bekanntermaßen guet, wenn der Ziel-Eigenzustand dünn besetzt isch: Die Wellenfunktion wird von aner Menge von Basiszuständ getrage, deren Größe ned exponenziell mit der Problemgröße wachst.
Quantenchemie
Die Eigenschafte von Moleküle werde weitgehend durch die Struktur der Elektronen in ihne bestimmt. Als fermionische Teilche könne Elektronen mit amm mathematische Formalismus, Zweitquantisierung genannt, beschriebe werde. Die Idee isch, dass es a Anzahl von Orbitale gibt, von dene jedes entweder leer isch oder von amm Fermion besetzt wird. A System von Orbitale wird durch an Satz fermionischer Vernichtungsoperatore beschriebe, die die fermionische Antikommutatorrelationen erfülle:
Der adjungierte Operator wird Erzeugungsoperator g'nannt.
Bis jetzt hat unsere Darstellung den Spin ned berücksichtigt, der a fundamentale Eigenschaft von Fermione isch. Beim Berücksichtige vom Spin komme die Orbitale in Paare vor, Raumorbitale g'nannt. Jedes Raumorbital besteht aus zwoi Spinorbitale, von dene ois mit "Spin-" und ois mit "Spin-" bezeichnet wird. Mir schreibe dann für den Vernichtungsoperator, der mit dem Spinorbital mit Spin () im Raumorbital assoziiert isch. Wenn mir als Anzahl der Raumorbitale nemme, gibt's insgesamt Spinorbitale. Der Hilbert-Raum von dem System wird von orthonormale Basisvektore aufgespannt, die mit zweiteilige Bitstrings bezeichnet werde: .
Der Hamiltonian vomme molekulare System ka g'schriebe werde als
wobei die und komplexe Zahle sind, die molekulare Integrale g'nannt werde und aus der Spezifikation vom Molekül mit amm Computerprogramm berechnet werde könne. In dem Tutorial berechne mir die Integrale mit em Softwarepaket PySCF.
Für Details darüber, wie der molekulare Hamiltonian hergeleitet wird, konsultiere Se a Lehrbuch zur Quantenchemie (zum Beispiel Modern Quantum Chemistry von Szabo und Ostlund). Für a übergeordnete Erklärung, wie Quantenchemieprobleme auf Quantencomputer abgebildet werde, schaut Se Euch die Vorlesung Mapping Problems to Qubits von der Qiskit Global Summer School 2024 a.
Local Unitary Cluster Jastrow (LUCJ) Ansatz
SQD braucht an Quantenschaltkreis-Ansatz, aus dem Stichproben g'zoge werde könne. In dem Tutorial verwende mir den Local Unitary Cluster Jastrow (LUCJ) Ansatz wege seiner Kombination aus physikalischer Motivation und Hardware-Freundlichkeit.
Der LUCJ-Ansatz isch a spezialisierte Form vom allgemeine Unitary Cluster Jastrow (UCJ) Ansatz, der die Form hat
wobei a Referenzzustand isch, oft der Hartree-Fock-Zustand, und die und die Form hend
wobei mir den Teilchenzahloperator definiert hend
Der Operator isch a Orbitalrotation, die mit bekannte Algorithme in linearer Tiefe und unter Verwendung linearer Konnektivität implementiert werde ka.
Die Implementierung vom Term vom UCJ-Ansatz erfordert entweder vollständige Konnektivität oder die Verwendung vomme fermionische Swap-Netzwerk, was für verrauschte präfehlertolerante Quantenprozessore mit begrenzter Konnektivität a Herausforderung isch. Die Idee vom lokale UCJ-Ansatz isch, Dünnbesetztheitsbedingunge auf die - und -Matrize aufz'erlege, die's ermögliche, sie in konstanter Tiefe auf Qubit-Topologien mit begrenzter Konnektivität z'implementiere. Die Bedingunge werde durch a Liste von Indizes spezifiziert, die a'zeige, welche Matrixeinträg im obere Dreieck von null verschieden sei dürfe (da die Matrize symmetrisch sind, muss nur des obere Dreieck spezifiziert werde). Diese Indizes könne als Orbitalpaare interpretiert werde, die mitanand interagiere dürfe.
Betrachte Se als Beispiel a quadratische Gitter-Qubit-Topologie. Mir könne die - und -Orbitale in parallele Linien auf dem Gitter platziere, wobei Verbindunge zwischen denne Linien "Sprossen" vonnere Leiterform bilde, wie folgt:

Bei dem Setup sind Orbitale mit demselbe Spin mit aner Linientopologie verbunde, während Orbitale mit unterschiedliche Spin verbunde sind, wenn sie dasselbe Raumorbital teile. Des ergibt die folgende Indexbedingunge für die -Matrize:
Mit andere Wort: Wenn die -Matrize nur an denne angegebene Indizes im obere Dreieck von null verschieden sind, ka der Term auf aner quadratische Topologie ohne Verwendung von Swap-Gates in konstanter Tiefe implementiert werde. Natürlich macht des Auferlege solche Bedingunge auf den Ansatz ihn weniger ausdrucksstark, sodass möglicherweise mehr Ansatz-Wiederholunge nötig sind.
Die IBM® Hardware hat a Heavy-Hex-Gitter-Qubit-Topologie, in welchem Fall mir a "Zickzack"-Muster verwende könne, des unten dargestellt isch. In dem Muster werde Orbitale mit demselbe Spin auf Qubits mit aner Linientopologie abgebildet (rote und blaue Kreise), und a Verbindung zwischen Orbitale unterschiedliche Spins isch an jedem 4. Raumorbital vorhande, wobei die Verbindung durch a Ancilla-Qubit (violette Kreise) ermöglicht wird. In dem Fall sind die Indexbedingunge
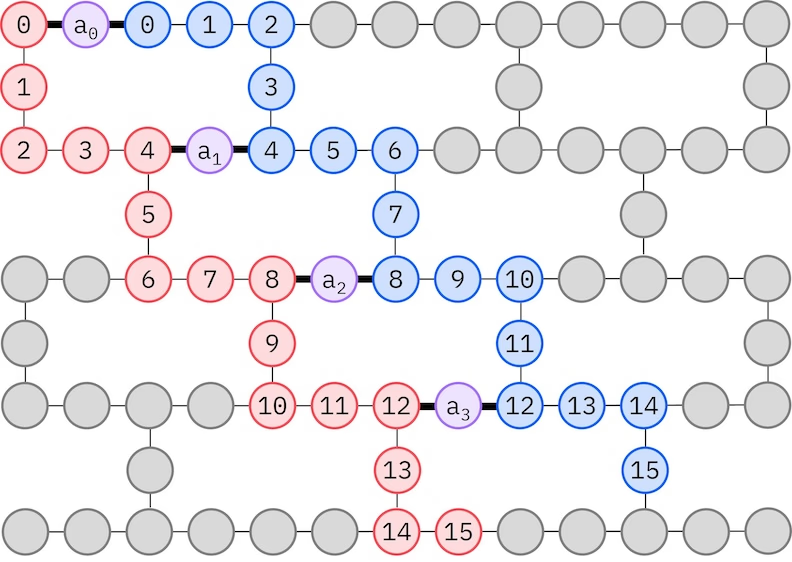
Selbstkonsistente Konfigurationswiederherstellung
Des selbstkonsistente Konfigurationswiederherstellungsverfahre isch dazu ausgelegt, so viel Signal wie möglich aus verrauschte Quantenstichproben raus z'hole. Da der molekulare Hamiltonian die Teilchenzahl und Spin-Z erhält, isch's sinnvoll, an Schaltkreis-Ansatz z'wähle, der diese Symmetriene au erhält. Wenn er auf den Hartree-Fock-Zustand a'g'wendet wird, hat der resultierende Zustand im rauschfreie Fall a feste Teilchenzahl und Spin-Z. Dohero sollte die Spin-- und Spin--Hälften von jedem aus dem Zustand g'sampelte Bitstring dasselbe Hamming-Gewicht wie im Hartree-Fock-Zustand hend. Wege dem Vorhandensein von Rauschen in aktuelle Quantenprozessore werde manche g'messene Bitstrings diese Eigenschaft verletze. A einfache Form der Postselektion würde diese Bitstrings verwerfe, aber des isch verschwenderisch, weil diese Bitstrings vielleicht no a bissle Signal enthalte. Des selbstkonsistente Wiederherstellungsverfahre versucht, a Teil von dem Signal in der Nachbearbeitung wiederherzustelle. Des Verfahre isch iterativ und braucht als Eingabe a Schätzung der durchschnittliche Besetzunge von jedem Orbital im Grundzustand, die zuerst aus denne Rohstichproben berechnet wird. Des Verfahre wird in aner Schleife ausgeführt, und jede Iteration hat die folgende Schritt:
- Für jeden Bitstring, der die spezifizierte Symmetriene verletzt, flippe Se seine Bits mit amm probabilistischen Verfahre, des dazu ausgelegt isch, den Bitstring näher an die aktuelle Schätzung der durchschnittliche Orbitalbesetzunge z'bringe, um an neue Bitstring z'erhalte.
- Sammle Se alle alte und neue Bitstrings, die die Symmetriene erfülle, und entnehme Se Teilmenge vonner im Voraus g'wählte feste Größe.
- Für jede Teilmenge von Bitstrings projiziere Se den Hamiltonian in den durch die entsprechende Basisvektore aufgespannte Unterraum (schaue Se in den vorige Abschnitt für a Beschreibung von denne Basisvektore) und berechne Se a Grundzustandsschätzung vom projizierten Hamiltonian auf amm klassische Computer.
- Aktualisiere Se die Schätzung der durchschnittliche Orbitalbesetzunge mit der Grundzustandsschätzung mit der niedrigste Energie.
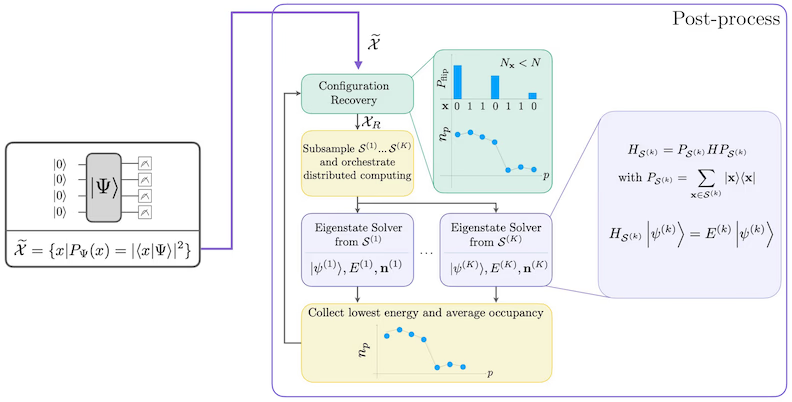
SQD-Workflow-Diagramm
Der SQD-Workflow isch im folgende Diagramm dargestellt:
Anforderunge
Stellt Se vor dem Beginne von dem Tutorial sicher, dass Se Folgendes installiert hend:
- Qiskit SDK v1.0 oder höher, mit Visualisierungs-Unterstützung
- Qiskit Runtime v0.22 oder höher (
pip install qiskit-ibm-runtime) - SQD Qiskit Addon v0.11 oder höher (
pip install qiskit-addon-sqd) - ffsim v0.0.58 oder höher (
pip install ffsim)
Setup
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime rustworkx
import pyscf
import pyscf.cc
import pyscf.mcscf
import ffsim
import numpy as np
import matplotlib.pyplot as plt
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
from qiskit_ibm_runtime import QiskitRuntimeService
from qiskit_ibm_runtime import SamplerV2 as Sampler
Schritt 1: Klassische Eingabe auf a Quantenproblem abbilden
In dem Tutorial finde mir a Approximation vom Grundzustand vom Molekül im Gleichgewicht im cc-pVDZ-Basissatz. Zuerst spezifiziere mir des Molekül und seine Eigenschafte.
# Specify molecule properties
open_shell = False
spin_sq = 0
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="cc-pvdz",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
num_orbitals = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
num_elec_a = (n_electrons + mol.spin) // 2
num_elec_b = (n_electrons - mol.spin) // 2
cas = pyscf.mcscf.CASCI(scf, num_orbitals, (num_elec_a, num_elec_b))
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), num_orbitals)
# Store reference energy from SCI calculation performed separately
exact_energy = -109.22690201485733
converged SCF energy = -108.929838385609
Vor dem Konstruiere vom LUCJ-Ansatz-Schaltkreis führe mir zunächst a CCSD-Berechnung in der folgende Code-Zelle durch. Die - und -Amplituden aus der Berechnung werde verwendet, um die Parameter vom Ansatz z'initialisiere.
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(
scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]
).run()
t1 = ccsd.t1
t2 = ccsd.t2
E(CCSD) = -109.2177884185543 E_corr = -0.2879500329450045
Jetzt verwende mir ffsim, um den Ansatz-Schaltkreis z'erstelle. Da unser Molekül an geschlossenschalige Hartree-Fock-Zustand hat, verwende mir die spin-balancierte Variante vom UCJ-Ansatz, UCJOpSpinBalanced. Mir übergebe Wechselwirkungspaare, die für a Heavy-Hex-Gitter-Qubit-Topologie geeignet sind (schaue Se in den Hintergrundabschnitt zum LUCJ-Ansatz). Mir setze optimize=True in der from_t_amplitudes-Methode, um die "komprimierte" Doppelfaktorisierung der -Amplituden z'ermögliche (schaue Se The local unitary cluster Jastrow (LUCJ) ansatz in der ffsim-Dokumentation für Details).
n_reps = 1
alpha_alpha_indices = [(p, p + 1) for p in range(num_orbitals - 1)]
alpha_beta_indices = [(p, p) for p in range(0, num_orbitals, 4)]
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(alpha_alpha_indices, alpha_beta_indices),
# Setting optimize=True enables the "compressed" factorization
optimize=True,
# Limit the number of optimization iterations to prevent the code cell from running
# too long. Removing this line may improve results.
options=dict(maxiter=1000),
)
nelec = (num_elec_a, num_elec_b)
# create an empty quantum circuit
qubits = QuantumRegister(2 * num_orbitals, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(num_orbitals, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
Schritt 2: Problem für Quantenhardware-Ausführung optimiere
Als Nächstes optimiere mir den Schaltkreis für a Ziel-Hardware.
service = QiskitRuntimeService()
backend = service.least_busy(
operational=True, simulator=False, min_num_qubits=133
)
print(f"Using backend {backend.name}")
Using backend ibm_fez
Mir empfehle die folgende Schritt, um den Ansatz z'optimiere und hardware-kompatibel z'mache.
- Wähle Se physikalische Qubits (
initial_layout) aus der Ziel-Hardware aus, die dem zuvor beschriebene "Zickzack"-Muster entspreche. Des Anlege von Qubits in dem Muster führt zu amm effizienten hardware-kompatible Schaltkreis mit weniger Gates. Do füge mir Code ein, um die Auswahl vom "Zickzack"-Muster z'automatisiere, der a Bewertungsheuristik verwendet, um die mit dem ausgewählte Layout verbundene Fehler z'minimiere. - Generiere Se an gestuften Pass-Manager mit der Funktion generate_preset_pass_manager von Qiskit mit Ihrer Wahl von
backendundinitial_layout. - Setze Se die
pre_init-Stufe von Ihrem gestuften Pass-Manager aufffsim.qiskit.PRE_INIT.ffsim.qiskit.PRE_INITenthält Qiskit-Transpiler-Pässe, die Gates in Orbitalrotationen zerleget und dann die Orbitalrotationen zusammenführet, was zu weniger Gates im endgültige Schaltkreis führt. - Führe Se den Pass-Manager auf Ihrem Schaltkreis aus.
Code für automatisierte Auswahl vom "Zickzack"-Layout
from typing import Sequence
import rustworkx
from qiskit.providers import BackendV2
from rustworkx import NoEdgeBetweenNodes, PyGraph
IBM_TWO_Q_GATES = {"cx", "ecr", "cz"}
def create_linear_chains(num_orbitals: int) -> PyGraph:
"""In zig-zag layout, there are two linear chains (with connecting qubits between
the chains). This function creates those two linear chains: a rustworkx PyGraph
with two disconnected linear chains. Each chain contains `num_orbitals` number
of nodes, that is, in the final graph there are `2 * num_orbitals` number of nodes.
Args:
num_orbitals (int): Number orbitals or nodes in each linear chain. They are
also known as alpha-alpha interaction qubits.
Returns:
A rustworkx.PyGraph with two disconnected linear chains each with `num_orbitals`
number of nodes.
"""
G = rustworkx.PyGraph()
for n in range(num_orbitals):
G.add_node(n)
for n in range(num_orbitals - 1):
G.add_edge(n, n + 1, None)
for n in range(num_orbitals, 2 * num_orbitals):
G.add_node(n)
for n in range(num_orbitals, 2 * num_orbitals - 1):
G.add_edge(n, n + 1, None)
return G
def create_lucj_zigzag_layout(
num_orbitals: int, backend_coupling_graph: PyGraph
) -> tuple[PyGraph, int]:
"""This function creates the complete zigzag graph that 'can be mapped' to an IBM QPU with
heavy-hex connectivity (the zigzag must be an isomorphic sub-graph to the QPU/backend
coupling graph for it to be mapped).
The zigzag pattern includes both linear chains (alpha-alpha interactions) and connecting
qubits between the linear chains (alpha-beta interactions).
Args:
num_orbitals (int): Number of orbitals, that is, number of nodes in each alpha-alpha linear chain.
backend_coupling_graph (PyGraph): The coupling graph of the backend on which the LUCJ ansatz
will be mapped and run. This function takes the coupling graph as a undirected
`rustworkx.PyGraph` where there is only one 'undirected' edge between two nodes,
that is, qubits. Usually, the coupling graph of a IBM backend is directed (for example, Eagle devices
such as ibm_brisbane) or may have two edges between two nodes (for example, Heron `ibm_torino`).
A user needs to be make such graphs undirected and/or remove duplicate edges to make them
compatible with this function.
Returns:
G_new (PyGraph): The graph with IBM backend compliant zigzag pattern.
num_alpha_beta_qubits (int): Number of connecting qubits between the linear chains
in the zigzag pattern. While we want as many connecting (alpha-beta) qubits between
the linear (alpha-alpha) chains, we cannot accommodate all due to qubit and connectivity
constraints of backends. This is the maximum number of connecting qubits the zigzag pattern
can have while being backend compliant (that is, isomorphic to backend coupling graph).
"""
isomorphic = False
G = create_linear_chains(num_orbitals=num_orbitals)
num_iters = num_orbitals
while not isomorphic:
G_new = G.copy()
num_alpha_beta_qubits = 0
for n in range(num_iters):
if n % 4 == 0:
new_node = 2 * num_orbitals + num_alpha_beta_qubits
G_new.add_node(new_node)
G_new.add_edge(n, new_node, None)
G_new.add_edge(new_node, n + num_orbitals, None)
num_alpha_beta_qubits = num_alpha_beta_qubits + 1
isomorphic = rustworkx.is_subgraph_isomorphic(
backend_coupling_graph, G_new
)
num_iters -= 1
return G_new, num_alpha_beta_qubits
def lightweight_layout_error_scoring(
backend: BackendV2,
virtual_edges: Sequence[Sequence[int]],
physical_layouts: Sequence[int],
two_q_gate_name: str,
) -> list[list[list[int], float]]:
"""Lightweight and heuristic function to score isomorphic layouts. There can be many zigzag patterns,
each with different set of physical qubits, that can be mapped to a backend. Some of them may
include less noise qubits and couplings than others. This function computes a simple error score
for each such layout. It sums up 2Q gate error for all couplings in the zigzag pattern (layout) and
measurement of errors of physical qubits in the layout to compute the error score.
Note:
This lightweight scoring can be refined using concepts such as mapomatic.
Args:
backend (BackendV2): A backend.
virtual_edges (Sequence[Sequence[int]]): Edges in the device compliant zigzag pattern where
nodes are numbered from 0 to (2 * num_orbitals + num_alpha_beta_qubits).
physical_layouts (Sequence[int]): All physical layouts of the zigzag pattern that are isomorphic
to each other and to the larger backend coupling map.
two_q_gate_name (str): The name of the two-qubit gate of the backend. The name is used for fetching
two-qubit gate error from backend properties.
Returns:
scores (list): A list of lists where each sublist contains two items. First item is the layout, and
second item is a float representing error score of the layout. The layouts in the `scores` are
sorted in the ascending order of error score.
"""
props = backend.properties()
scores = []
for layout in physical_layouts:
total_2q_error = 0
for edge in virtual_edges:
physical_edge = (layout[edge[0]], layout[edge[1]])
try:
ge = props.gate_error(two_q_gate_name, physical_edge)
except Exception:
ge = props.gate_error(two_q_gate_name, physical_edge[::-1])
total_2q_error += ge
total_measurement_error = 0
for qubit in layout:
meas_error = props.readout_error(qubit)
total_measurement_error += meas_error
scores.append([layout, total_2q_error + total_measurement_error])
return sorted(scores, key=lambda x: x[1])
def _make_backend_cmap_pygraph(backend: BackendV2) -> PyGraph:
graph = backend.coupling_map.graph
if not graph.is_symmetric():
graph.make_symmetric()
backend_coupling_graph = graph.to_undirected()
edge_list = backend_coupling_graph.edge_list()
removed_edge = []
for edge in edge_list:
if set(edge) in removed_edge:
continue
try:
backend_coupling_graph.remove_edge(edge[0], edge[1])
removed_edge.append(set(edge))
except NoEdgeBetweenNodes:
pass
return backend_coupling_graph
def get_zigzag_physical_layout(
num_orbitals: int, backend: BackendV2, score_layouts: bool = True
) -> tuple[list[int], int]:
"""The main function that generates the zigzag pattern with physical qubits that can be used
as an `intial_layout` in a preset passmanager/transpiler.
Args:
num_orbitals (int): Number of orbitals.
backend (BackendV2): A backend.
score_layouts (bool): Optional. If `True`, it uses the `lightweight_layout_error_scoring`
function to score the isomorphic layouts and returns the layout with less erroneous qubits.
If `False`, returns the first isomorphic subgraph.
Returns:
A tuple of device compliant layout (list[int]) with zigzag pattern and an int representing
number of alpha-beta-interactions.
"""
backend_coupling_graph = _make_backend_cmap_pygraph(backend=backend)
G, num_alpha_beta_qubits = create_lucj_zigzag_layout(
num_orbitals=num_orbitals,
backend_coupling_graph=backend_coupling_graph,
)
isomorphic_mappings = rustworkx.vf2_mapping(
backend_coupling_graph, G, subgraph=True
)
isomorphic_mappings = list(isomorphic_mappings)
edges = list(G.edge_list())
layouts = []
for mapping in isomorphic_mappings:
initial_layout = [None] * (2 * num_orbitals + num_alpha_beta_qubits)
for key, value in mapping.items():
initial_layout[value] = key
layouts.append(initial_layout)
two_q_gate_name = IBM_TWO_Q_GATES.intersection(
backend.configuration().basis_gates
).pop()
if score_layouts:
scores = lightweight_layout_error_scoring(
backend=backend,
virtual_edges=edges,
physical_layouts=layouts,
two_q_gate_name=two_q_gate_name,
)
return scores[0][0][:-num_alpha_beta_qubits], num_alpha_beta_qubits
return layouts[0][:-num_alpha_beta_qubits], num_alpha_beta_qubits
initial_layout, _ = get_zigzag_physical_layout(num_orbitals, backend=backend)
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend, initial_layout=initial_layout
)
# without PRE_INIT passes
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/o pre-init passes): {isa_circuit.count_ops()}")
# with PRE_INIT passes
# We will use the circuit generated by this pass manager for hardware execution
pass_manager.pre_init = ffsim.qiskit.PRE_INIT
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/ pre-init passes): {isa_circuit.count_ops()}")
Gate counts (w/o pre-init passes): OrderedDict({'rz': 12438, 'sx': 12169, 'cz': 3986, 'x': 573, 'measure': 52, 'barrier': 1})
Gate counts (w/ pre-init passes): OrderedDict({'sx': 7059, 'rz': 6962, 'cz': 1876, 'measure': 52, 'x': 35, 'barrier': 1})
Schritt 3: Ausführe mit Qiskit-Primitive
Nach der Optimierung vom Schaltkreis für die Hardware-Ausführung sind mir bereit, ihn auf der Ziel-Hardware auszuführe und Stichproben für die Grundzustandsenergieabschätzung z'sammle. Da mir nur an Schaltkreis hend, verwende mir den Job-Ausführungsmodus von Qiskit Runtime und führe unsere Schaltkreis aus.
sampler = Sampler(mode=backend)
job = sampler.run([isa_circuit], shots=100_000)
primitive_result = job.result()
pub_result = primitive_result[0]
Schritt 4: Nachbearbeitung und Rückgabe vom Ergebnis im g'wünschte klassische Format
Jetzt schätze mir die Grundzustandsenergie vom Hamiltonian mit der Funktion diagonalize_fermionic_hamiltonian. Diese Funktion führt des selbstkonsistente Konfigurationswiederherstellungsverfahre aus, um die verrauschte Quantenstichproben iterativ z'verfeinere und die Energieabschätzung z'verbessere. Mir übergebe a Callback-Funktion, damit mir die Zwischenergebnisse für a spätere Analyse speichere könne. Schaue Se in die API-Dokumentation für Erklärunge der Argumente von diagonalize_fermionic_hamiltonian.
from functools import partial
from qiskit_addon_sqd.fermion import (
SCIResult,
diagonalize_fermionic_hamiltonian,
solve_sci_batch,
)
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 3
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the sci_solver argument
# in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy + nuclear_repulsion_energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
pub_result.data.meas,
samples_per_batch=samples_per_batch,
norb=num_orbitals,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
carryover_threshold=carryover_threshold,
callback=callback,
seed=12345,
)
Iteration 1
Subsample 0
Energy: -108.97781410104506
Subspace dimension: 28561
Subsample 1
Energy: -108.97781410104506
Subspace dimension: 28561
Subsample 2
Energy: -108.97781410104506
Subspace dimension: 28561
Iteration 2
Subsample 0
Energy: -109.05150860754739
Subspace dimension: 287296
Subsample 1
Energy: -109.08152283263908
Subspace dimension: 264196
Subsample 2
Energy: -109.11636893067873
Subspace dimension: 284089
Iteration 3
Subsample 0
Energy: -109.15878555367885
Subspace dimension: 429025
Subsample 1
Energy: -109.16462831275786
Subspace dimension: 473344
Subsample 2
Energy: -109.15895026995382
Subspace dimension: 435600
Iteration 4
Subsample 0
Energy: -109.17784051223317
Subspace dimension: 622521
Subsample 1
Energy: -109.1774651326829
Subspace dimension: 657721
Subsample 2
Energy: -109.18085212360191
Subspace dimension: 617796
Iteration 5
Subsample 0
Energy: -109.18636242518915
Subspace dimension: 815409
Subsample 1
Energy: -109.18451014767518
Subspace dimension: 837225
Subsample 2
Energy: -109.18333728638888
Subspace dimension: 857476
Visualisierung der Ergebnisse
Der erste Plot zeigt, dass mir nach a paar Iterationen die Grundzustandsenergie innerhalb von ~41 mH schätze (chemische Genauigkeit wird typischerweise als 1 kcal/mol 1.6 mH akzeptiert). Die Energie ka verbessert werde, indem mehr Iterationen der Konfigurationswiederherstellung erlaubt werde oder die Anzahl der Stichproben pro Batch erhöht wird.
Der zweite Plot zeigt die durchschnittliche Besetzung von jedem Raumorbital nach der letzte Iteration. Mir könne sehe, dass sowohl die Spin-Up- als au die Spin-Down-Elektronen die erste fünf Orbitale mit hoher Wahrscheinlichkeit in unsere Lösungen besetze.
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - exact_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(
y=chem_accuracy,
color="#BF5700",
linestyle="--",
label="chemical accuracy",
)
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
plt.tight_layout()
plt.show()

Tutorial-Umfrage
Bitte nehme Se an dere kurze Umfrage teil, um Feedback zu dem Tutorial z'gebe. Ihre Einblicke helfe uns, unsere Inhaltsangebote und Benutzererfahrung z'verbessere.